
To watch the presentation by Dr. Bart P. Leroy at EURETINA, please click on the link at the end of this article.

IRDs are a clinically and genetically heterogeneous group of conditions.10 Over 300 genes have been identified as being responsible for causing IRDs, defects in which can result in a wide, overlapping spectrum of conditions, including congenital night blindness, cone dysfunction syndromes, and retinitis pigmentosa.11 These clinical phenotypes can each be caused by a number of different mutations.12 For example, 24 genes are currently known to be involved in Leber congenital amaurosis and early onset retinal dystrophy, including RPE65.13 With the advent of gene therapies specific to an individual gene or even an individual mutation,14,15 the classification of IRDs is now moving away from a definition based on the clinical condition towards a diagnosis being based on the specific underlying gene mutation. “Gene therapy is gene specific, and there’s no margin for error, so we need to know that what we are doing is right,” said Dr. Leroy.
For this reason, genotyping is crucial in any patient suspected to have an IRD. Currently, the nature of the patient referral pathway means that it can take a number of years for a patient with an IRD to reach a super-specialist center where they can receive a definitive molecular diagnosis of their condition. “Ophthalmologists have a duty to recognize such disease and then not delay sending a patient for review by a specialist. Information-sharing and effective interactions between clinicians and specialists are essential,” said Dr. Leroy. To help streamline the patient referral process and improve the coordination of care for patients with rare eye diseases, the European Reference Network for Rare Eye Disease (ERN-EYE), a network of 29 health care providers in 13 European Union member states, has been created. The aim of ERN-EYE is to ensure patients receive an accurate and timely diagnosis, high-quality care, access to innovation, and the opportunity to be involved in clinical trials.16 Within ERN-EYE, rare eye disease experts integrate with their European peers and form collaborations with patient groups in order to contribute to improving patient care for individuals with IRDs. A European Union initiative, such ERNs exist for multiple types of rare diseases.
In an ocular genetics evaluation for a patient with a suspected IRD, it’s important to ask the right questions about the nature of the visual complaints and the time of onset of symptoms, using language the that patient can understand. The evaluation will also entail the drawing of a pedigree, and the clinical diagnosis should be supported with specialized imaging, psychophysics, and electrophysiology (sometimes termed ‘deep phenotyping’). Finally, and importantly, the diagnosis must be confirmed with molecular testing. “Often this type of detailed evaluation is not possible in a busy retinal practice, so please do refer your patients to a specialist,” said Dr. Leroy.
Techniques for detecting mutations have evolved within the past decade from testing for known mutations using Sanger sequencing to next generation sequencing in which the whole exome is read in a high-throughput process (Figure 5).17 Targeted gene testing is still performed today, but now uses panels of genes that capture exons of multiple IRD genes. This is a highly sensitive technique, which is also flexible, easy, and cost-effective.18 With whole exome sequencing, the entire exome of an individual can be sequenced to identify causative mutations, with one such sequencing procedure revealing up to 100,000 variants. Comparative genome hybridization can also be performed, which allows the detection of copy number variations.19 However, molecular analysis alone fails to identify a causative gene in about 40% of patients, with the proportion of patients having an unsolved outcome varying between conditions (Figure 6).
Images provided courtesy of Caroline Van Cauwenbergh, PhD.
Figure 5. Evolution of mutation detection techniques.
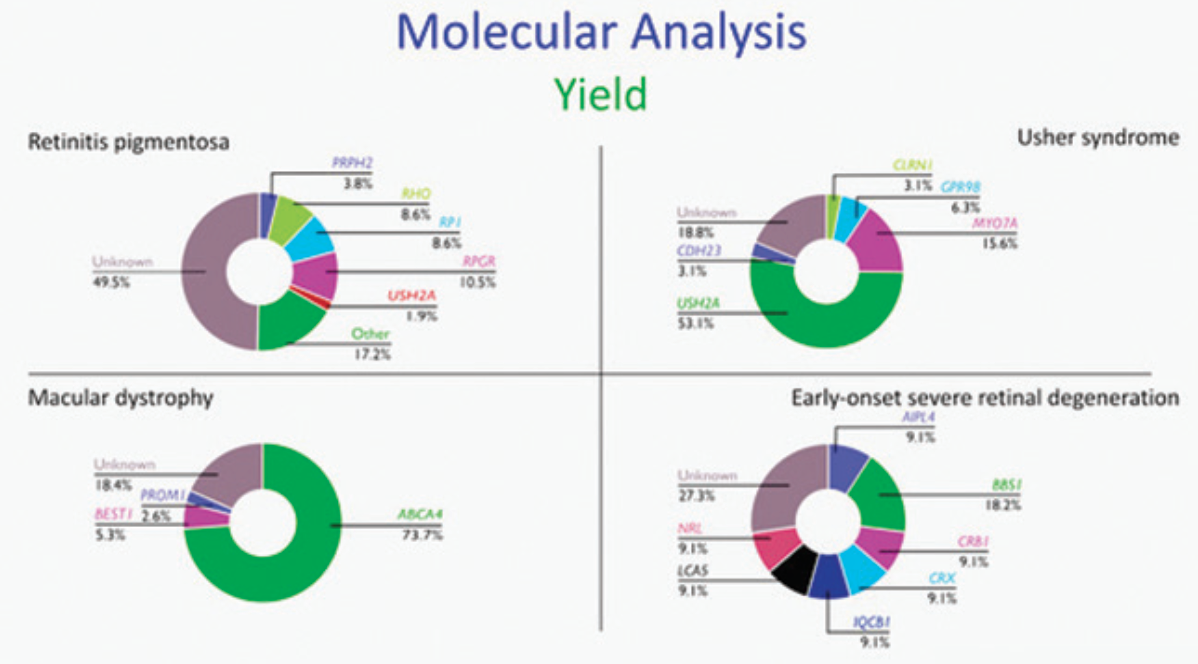
Images provided courtesy of Caroline Van Cauwenbergh, PhD.
Figure 6. Genetic variants identified in IRDs.
Genetic variation used to be classified as either a polymorphism (implying a change with no disease-bearing importance) or a disease-causing mutation. In 2015, the American College of Medical Genetics last updated the definition for clinical significance of any given sequence variant, which actually falls somewhere along a gradient from pathogenic to benign, and they recommended the use of specific standard terminology: ‘pathogenic,’ ‘likely pathogenic,’ ‘uncertain significance,’ ‘likely benign,’ and ‘benign’ (Figure 7).20 “It often takes a lot of work to determine how a variant should be classified, with treatment only possible for patients with pathogenic or likely pathogenic variants,” said Dr. Leroy.

Images provided courtesy of Caroline Van Cauwenbergh, PhD.
Figure 7. American Society for Molecular Genetics classification of sequence variants.
In the case of autosomal recessive disease, which requires two mutations (one on each allele), genetic testing of the patient’s parents (or if not available, their siblings or children) is vital. This is in order to unequivocally confirm that the two mutations reside on different alleles (i.e. recessive disease) rather than the two changes being carried on the same allele, which would mean that the gene was not the basis of the disease.
Genotyping is required to confirm eligibility for treatment with voretigene neparvovec as this gene therapy is only indicated for adult and pediatric patients with vision loss due to inherited retinal dystrophy caused by confirmed bi-allelic RPE65 mutations.21 It is also necessary for the patient to have sufficient viable retinal cells. In practice, this is determined by the IRD specialist checking for the presence of outer retinal cells on SD-OCT and the presence of at least light perception upon visual testing. Some additional measurement of visual function, such as FST, is also desirable.
To watch the presentation by Dr. Bart P. Leroy at EURETINA 2019, please click here.